Large Cycloparaphenylene Nanolassos Characterized with AIQM1

Alkyne-embedding [11]cycloparaphenylene ([11]CPPs) was functionalized with electron-donating, -neutral, and -withdrawing aryl substituents to yield a series of nanolassos via click chemistry. We used our state-of-the-art, artificial intelligence-enhanced quantum mechanical method 1 (AIQM1) to thoroughly analyze the electronic and photophysical properties of these compounds and their complexes with fullerenes. Out-of-the-box AIQM1 allowed performing fast and accurate calculations to better understand and explain the phenomena observed experimentally for these big systems with hundreds of atoms.
In our recent work, our collaborators functionalized strained alkyne-embedding [11]CPP with a series of electron-donating, -neutral, and -withdrawing aryl substituents through copper-free [3+2]azide-alkyne click chemistry. This functionalization resulted in lasso-shaped structures shown below.
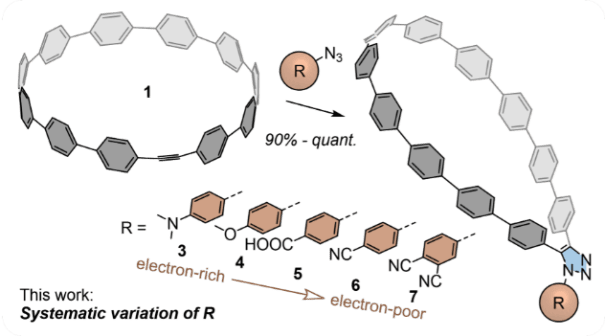
The collaborators also analyzed these molecular nanolassos by X-ray crystallography and their electronic and photophysical properties were probed with NMR spectroscopy, steady-state and transient absorption and emission spectroscopies as well as electrochemistry. The shape of the above compounds allows us to easily capture fullerenes and our study investigated nanolosso-fullerene complexes too.
Our group performed theoretical calculations to explain the nature of observed experimental phenomena. The nanolassos and their complexes with fullerenes are rather large with up to several hundreds of atoms, thus, it is computationally expensive to perform high-level calculations on them. Thus, to ensure the high quality of the calculations, we mostly used our AIQM1 approach which is both faster and more accurate than popular DFT approaches.
To verify the use of AIQM1 for compounds of this size, we compared AIQM1-optimized geometry with X-ray crystallographic structure of one of the compounds. As can be seen from the figure below, the root mean square displacement (RMSD) between two geometries is quite small (ca. 0.63 Å) with differences that may be also caused by the packing effects not considered in our gas-phase optimizations. For comparison, DFT optimization is both much more time-consuming (20 CPU-hours compared to 2 CPU-minutes at AIQM1) and not more accurate (RMSD of 0.66 Å).

Theoretical 13C NMR shifts calculated with DFT on AIQM1-optimized geometries allow to unambiguously assign experimentally observed peaks to specific atoms and show downfield shifts for the Cipso atoms with increasing electron-withdrawing properties of the substituents. AIQM1 calculations of oxidation and reduction properties of nanolassos not just followed the expected trend for substituents but also revealed the charge distribution upon oxidation and reduction, which ranges from full to partial electron/hole localization on the carbon loop/substituent.

One of the key observations which was challenging to explain is the solvent effect on the absorption and fluorescence of nanolassos: The spectra are slightly red-shifted in more polar solvent. One could guess that it may be due to the plausible intramolecular charge transfer, but our TDDFT calculations on the AIQM1-optimized geometries and subsequent one-electron density matrix (1TDM) analysis did not reveal any charge transfer between carbon loop and substitute and shows that excitations are localized on the carbon loop (Frenkel excitons). This puzzling phenomenon was explained by subtle but important effect of solvent on geometry in the ground state.

Turning to the complexation of nanolassos with fullerenes, we found that complexation with C70 is stronger than to C60as the oval shape of C70 is a better match to the lasso-shape of CPP derivatives. This is confirmed by our AIQM1 calculations and our collaborators’ experimental mass-spectrometry measurements. From the experiment, however, we could not judge how exactly is C70 aligned relative to the carbon loop: it could be in lying (L-) or standing (S-) or in half-lying, half-standing (M-). With AIQM1, we could easily analyze and find that the most stable configuration is lying, see the Figure below for geometries and binding energies.

Importantly, configuration interaction singles (CIS) calculations with AIQM1 allowed use to optimize geometries of free nanolassos and their complexes with the fullerenes in the first excited state and calculate fluorescence. Our AIQM1/CIS calculations showed that fluorescence of free nanolassos should be quenched upon complexation and this is indeed what was observed experimentally.
Our work clearly demonstrates that AI-enhanced quantum mechanical methods can be already routinely used to investigate complex phenomena in organic compounds with high accuracy and speed. Noteworthy, all above calculations were done with the out-of-the-box AIQM1 without any retraining, etc., i.e., as one would use any other typical quantum chemical methods.
AIQM1 is open-source and free to use with detailed tutorials on its use available at MLatom.com/AIQM1 and basic calculations with AIQM1 can be performed free of charge via a web browser using our MLatom@XACS cloud computing service.
Paper:
- Tobias A. Schaub*, Anna Zieleniewska, Ramandeep Kaur, Martin Minameyer, Wudi Yang, Christoph M. Schüßlbauer, Lina Zhang, Markus Freiberger, Lev N. Zakharov, Thomas Drewello, Pavlo O. Dral, Dirk Guldi, Ramesh Jasti*. Tunable Macrocyclic Polyparaphenylene Nanolassos via Copper‐Free Click Chemistry. Chem. Eur. J. 2023, in press.