MLatom 2: Introducing a Platform for Atomistic Machine Learning

We are happy to introduce MLatom 2: a major release of our integrative platform for user-friendly atomistic machine learning. It includes many more features and is further optimized for efficiency. Detailed overview of MLatom 2 is given in our contribution to the topical collection New Horizon in Computational Chemistry Software of the Topics in Current Chemistry.
Since its inception, the philosophy behind MLatom was to provide easy-to-use platform for routine atomistic simulations with machine learning in order to help new researchers in overcoming the entry barrier. Thus, the user only needs to specify the right keywords and prepare input data rather than to modify scripts (but you can, if you want). As such, it is also a useful tool for teaching courses including topics on machine learning in chemistry and one can for this use our tutorial and book chapter. The package now offers a wide range features required for atomistic machine learning:
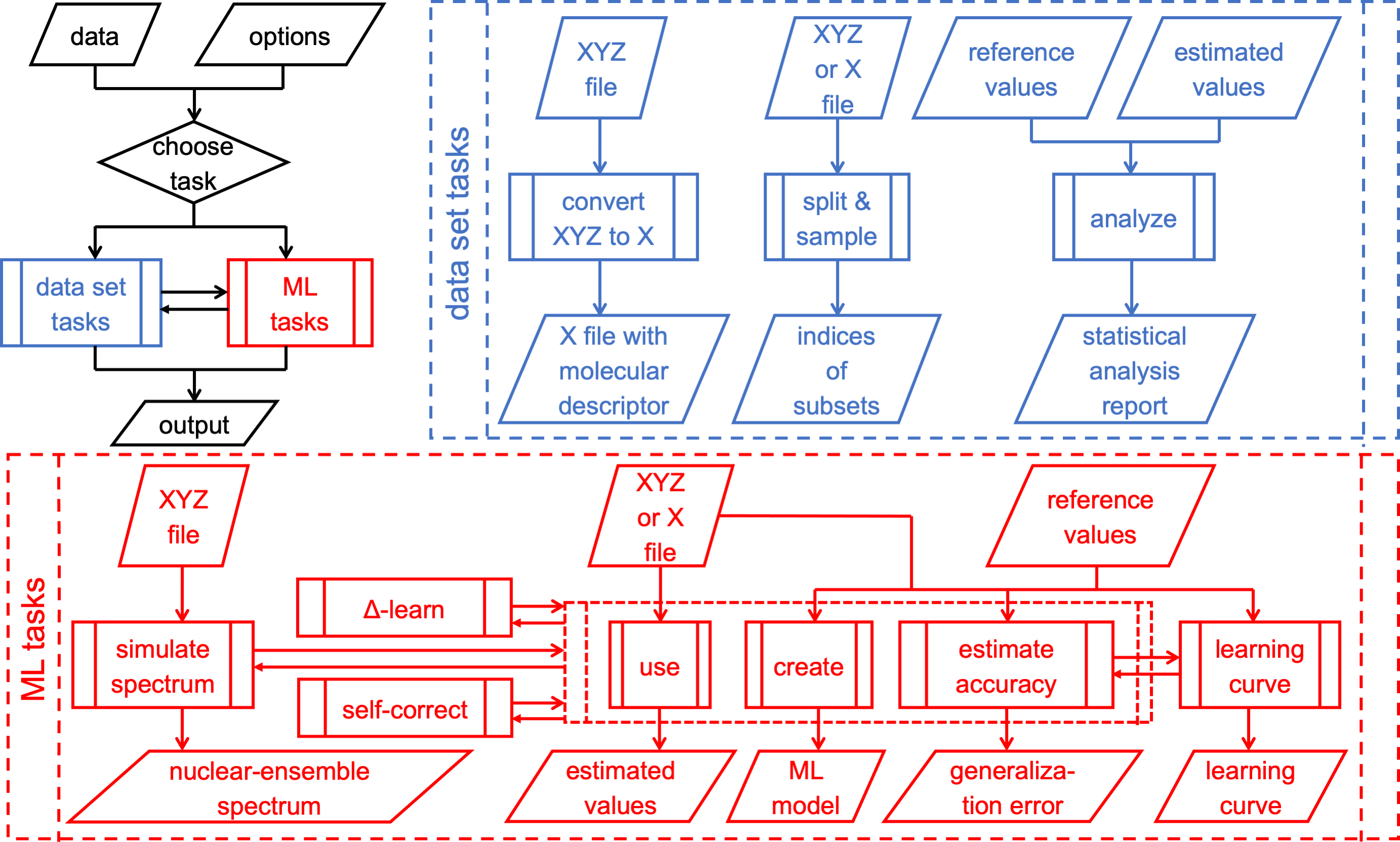
The core of MLatom is our own native implementations based on kernel ridge regression such as the KREG model, which can be used to accurately learn properties of different conformations of the same molecule (e.g. accurate potential energy surfaces including permutational invariance). Analytical gradients are also available for such models (co-implemented by Yi-Fan Hou). Higher-level operations such as Δ-learning and self-correction are also supported.
Now you can perform such multi-step tasks as automatically generating UV/vis absorption spectrum within machine learning-nuclear ensemble approach just by providing the initial geometry and specifying the quantum chemical level of theory for calculating frequencies and reference excited-state properties for training:
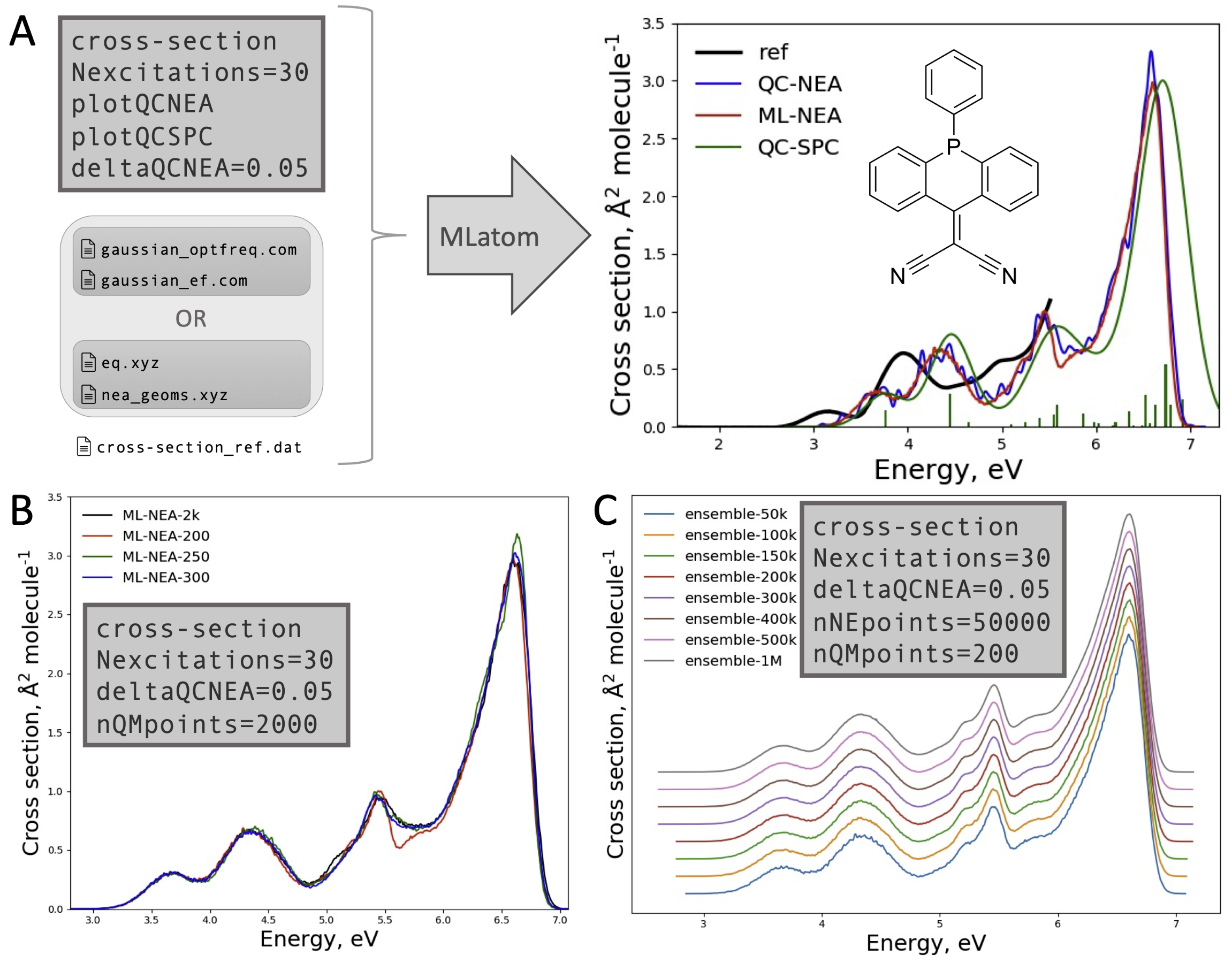
This operation was implemented by Bao-Xin Xue and is based on interfaces to third-party software such as Newton-X by Mario Barbatti et al. We are now lucky to have Mario’s group involved in further implementations in MLatom and thanks to the efforts of Max Pinheiro Jr from his group and Fuchun Ge with Jianxing Huang from Xiamen University, MLatom 2 features support to many popular, state-of-the-art machine learning potentials such as sGDML, GAP-SOAP, ANI, DeepPot-SE, and PhysNet through interfaces to the programs by developers of these methods (see the links):
- ANI (through TorchANI)
- DeepPot-SE and DPMD (through DeePMD-kit)
- GAP–SOAP (through GAP suite and QUIP)
- PhysNet (through PhysNet)
- sGDML (through sGDML)
Once these programs are installed, MLatom can be used to run calculations with any of the above model types by simply changing several keywords to specify the model type and this model specific keywords (we provide many default settings), so that the user does not need to learn the intricacies of using each of these packages, but can utilize their capabilities:

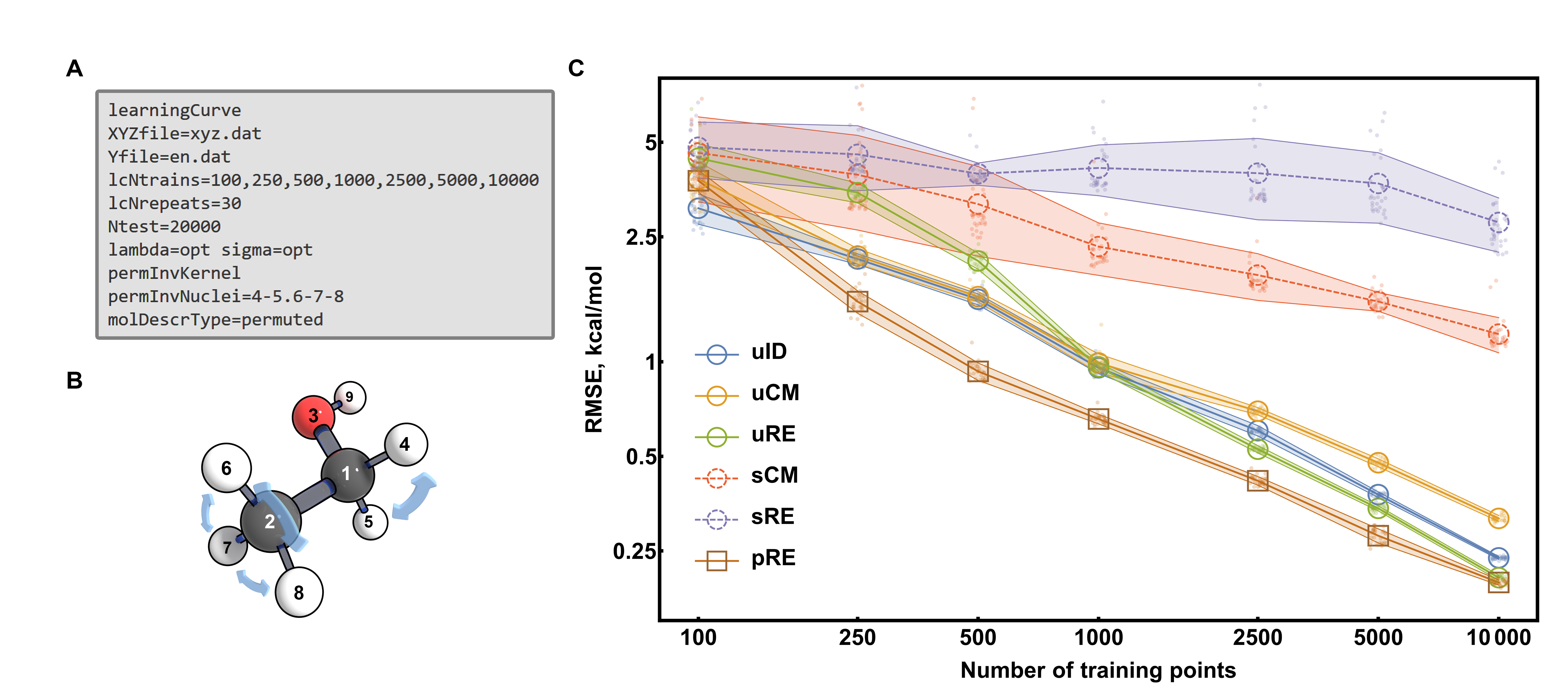
The above input shows an example of generating learning curves, which are very important to compare the performance of different models as comparisons based just on a single training set size may give wrong conclusions, as curves slopes and intercepts can be very different!
This is Fuchun’s implementation too, who also implemented of the interface to the hyperopt package enabling tuning hyperparameters of any generic model, e.g. for DeepPot-SE:
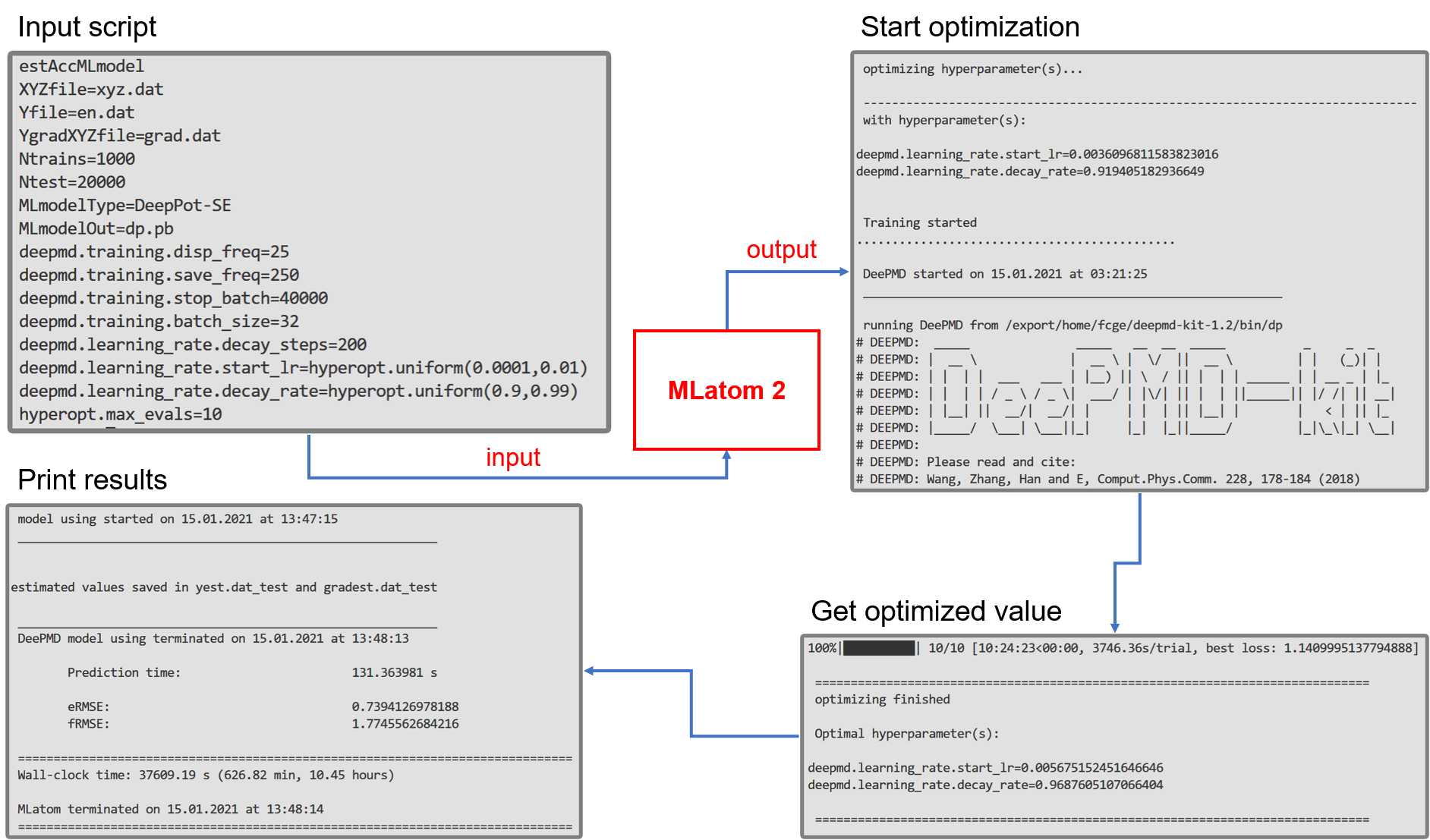
The modular structure of implementation of interfaces with Python allows for easy extension to new machine learning potentials – if you want to contribute please let us know.
The package can be either downloaded from the MLatom website or by running the command in your terminal on Linux:
python3 -m pip install -U MLatom
You may run this command from time to time to update the package for new features and bug fixes. You can also subscribe to the updates on MLatom website.
Stay tuned for future updates as more exciting features are coming!
- Pavlo O. Dral, Fuchun Ge, Bao-Xin Xue, Yi-Fan Hou, Max Pinheiro Jr, Jianxing Huang, Mario Barbatti, MLatom 2: An Integrative Platform for Atomistic Machine Learning. Top. Curr. Chem. 2021, 379, 27. DOI: 10.1007/s41061-021-00339-5. Open access; some figures are taken from this article.
I would like to learn computational chemistry