Correcting Differences with Machine Learning
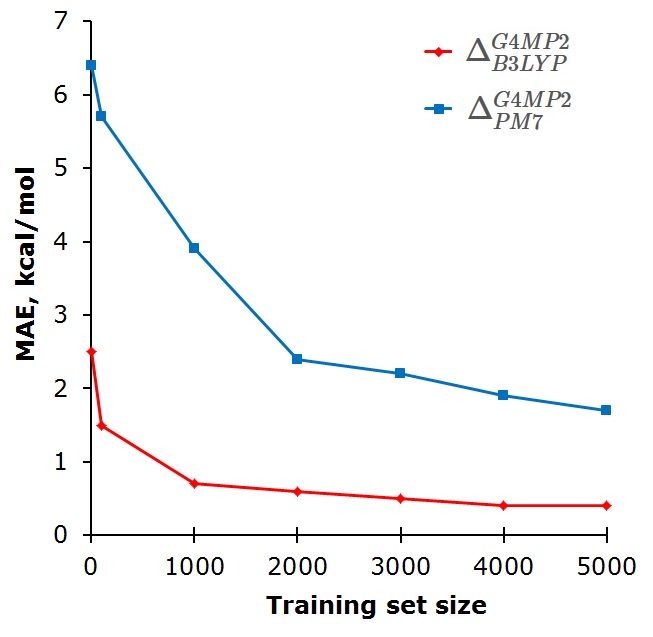
In our recent study, we propose using machine learning (ML) to correct differences in properties calculated at two quantum chemical (QC) methods with different accuracy.
In the Δ-ML approach ML model is trained on differences between some property \(P_t\) calculated at the target level of theory and property \(P’_b\) calculated at the baseline level of theory. Then this ML model is used to predict \(\Delta_b^t\) for out-of-sample molecules.
Such Δ-ML approach can be used for correcting errors in property calculated at less accurate, but faster QC method. After generating reference data for training set molecules at more accurate, but slower level of theory, training ML model is relatively fast and ML-corrections are computed for new molecules essentially for free.
The Δ-ML approach can be also used for correcting differences between different properties. For instance, B3LYP can be used to calculate atomization energies and then ML corrects them to obtain enthalpies of atomization at 298 K with chemical accuracy (relative to more accurate target line G4MP2 method).
The concepts behind the Δ-ML and the automatic parametrization technique (APT) published earlier differ fundamentally. The Δ-ML makes explicit on-top corrections to properties pre-calculated with less accurate QC methods, while the APT improves QC methods implicitly by correcting their parameters, which are then used to calculate required properties. Thus, one or another of these approaches may be more preferable for the problem at hand.
- Raghunathan Ramakrishnan, Pavlo O. Dral, Matthias Rupp, O. Anatole von Lilienfeld, Big Data meets Quantum Chemistry Approximations: The Δ-Machine Learning Approach. J. Chem. Theory Comput. 2015, Just Accepted Manuscript. DOI: 10.1021/acs.jctc.5b00099.
arXiv:1503.04987 [physics.chem-ph].
This Δ-ML approach was featured and described in detail in Computational Chemistry Highlights (Jan Jensen, Comp. Chem. Highlights, 2015.04.big)