Machine Learning in Chemistry
We are developing and using machine learning (ML) for improving and accelerating quantum chemical methods and dynamics. Based on our rich experience in working this field since 2013, we have offered a concise overview of the field in our Perspective Quantum Chemistry in the Age of Machine Learning pointing out the main directions and challenges.[9] We are also committed to educating students about how ML can assist in quantum chemical research and for this we have written a book chapter providing many examples in a tutorial way.[12]
Our research can be roughly split in three main directions covered on this page: 1) ML-enhanced quantum chemical methods, 2) ML of ground-state potentials, 3) ML of excited-state properties. An out-of-date list of our publications in this field is given at the bottom of the page. For this research, we also develop MLatom, which is our own package for easy-to-use, practical and efficient atomistic simulations with ML.[7][8][14] In addition, our data sets such as QM9 are freely available to the community.[1]
ML-enhanced quantum chemical methods
We develop ML methods to improve accuracy of quantum mechanical (QM) methods—DFT and especially semiempirical quantum chemical (SQC) methods—and use them for calculating various molecular properties with reasonable accuracy and low computational cost. The pinnacle of our research in this direction is AIQM1 (Artificial Intelligence-enhanced quantum mechanical method 1), which provides out-of-the-box predictions for organic molecules with accuracy approaching that of CCSD(T)/CBS for ground-state neutral species, but also transferable to other properties and species with good accuracy.[17]

Earlier, we have introduced two other general approaches. The first one is implicitly improving QM methods. We do it by correcting semiempirical Hamiltonian using ML and for this we introduced ML-automatic parameterization technique, (ML-APT).[2]
The second approach is improving explicitly QM methods by making on-top corrections with ML (Δ-ML technique).[3]
All these hybrid ML/QM methods break through the limited transferability of pure ML methods, which often have very inaccurate, unphysical behavior far outside their training domain.
ML of ground-state potentials

ML potentials is likely the most popular application of ML in quantum chemistry. There is so much research done in this area that it is easy to get lost in the sea of published literature and difficult to choose a good ML potential for your research. Thus, we provide guidelines in our work,[15] where we also compared the performance of many popular ML potentials. Among them, our KREG approach (kernel-ridge-regression with Gaussian kernel and RE descriptor with normalized inverse internuclear distances) turns out to be a good choice for fast and accurate training on energies.[4]
It is not enough to have a good ML potential for reducing the cost of quantum chemical calculations. Thus, we have proposed structure-based sampling and self-correcting machine learning (ML) for precise representation of molecular potential energy surfaces and calculating vibrational levels with spectroscopic accuracy (errors less than 1 cm−1 relative to the reference ab initio spectrum).[4] Structure-based sampling ensures that ML works most of the time in the interpolation regime, where it performs best. Our approach reduces the number of required quantum mechanical calculations by up to 90%. We have also generalized Δ-machine learning to semi-automatically define training sets for many quantum chemical corrections in order to build hierarchical machine learning (hML) potentials of high accuracy (this approach could reduce the required computational time up to 99%).[10]
ML of excited-state properties
Applying ML to excited-state properties is highly attractive due to staggering cost of calculating electronic excitations with quantum chemical methods. Thus, many methods have been suggested and we provide a Review of ML in excited-state research.[13]
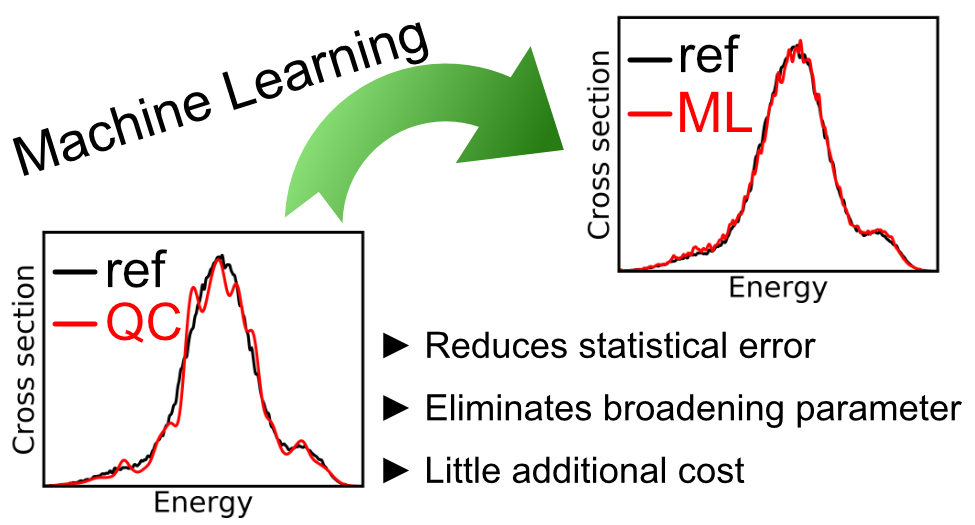
Our research contributions to the field include ML-nuclear ensemble approach for cost-efficient and high-precision simulation of absorption spectra[11] as well as studies on accelerating nonadiabatic excited-state dynamics[5][6] and quantum dissipative dynamics of open systems[16] with machine learning.

Publications
17. Peikun Zheng, Roman Zubatyuk, Wei Wu, Olexandr Isayev, Pavlo O. Dral, Artificial Intelligence-Enhanced Quantum Chemical Method with Broad Applicability, Nat. Commun., 2021, 12, 7022. DOI: 10.1038/s41467-021-27340-2.
16. Arif Ullah, Pavlo O. Dral. Speeding up quantum dissipative dynamics of open systems with kernel methods, New J. Phys., 2021, accepted. DOI: 10.1088/1367-2630/ac3261.
15. Max Pinheiro Jr, Fuchun Ge, Nicolas Ferré, Pavlo O. Dral, Mario Barbatti. Choosing the right molecular machine learning potential. Chem. Sci., 2021, accepted. DOI: 10.1039/D1SC03564A. (blog post)
14. Pavlo O. Dral, Fuchun Ge, Bao-Xin Xue, Yi-Fan Hou, Max Pinheiro Jr, Jianxing Huang, Mario Barbatti, MLatom 2: An Integrative Platform for Atomistic Machine Learning. Top. Curr. Chem. 2021, 379, 27. DOI: 10.1007/s41061-021-00339-5. (blog post)
13. Pavlo O. Dral, Mario Barbatti, Molecular excited states through a machine learning lens, Nat. Rev. Chem. 2021, 5, 388–405. DOI: 10.1038/s41570-021-00278-1. (blog post)
12. Pavlo O. Dral, Quantum Chemistry Assisted by Machine Learning. In Advances in Quantum Chemistry: Chemical Physics and Quantum Chemistry, Volume 81, 1st ed.; Kenneth Ruud, Erkki J. Brändas, Eds. Academic Press: 2020; Vol. 81. DOI: 10.1016/bs.aiq.2020.05.002. (blog post | online tutorial)
11. Bao-Xin Xue, Mario Barbatti, Pavlo O. Dral, Machine Learning for Absorption Cross Sections, J. Phys. Chem. A 2020, 124, 7199–7210. DOI: 10.1021/acs.jpca.0c05310.
10. Pavlo O. Dral, Alec Owens, Alexey Dral, Gábor Csányi, Hierarchical Machine Learning of Potential Energy Surfaces. J. Chem. Phys. 2020, 152, 204110. DOI: 10.1063/5.0006498.
9. Pavlo O. Dral, Quantum Chemistry in the Age of Machine Learning. J. Phys. Chem. Lett. 2020, 11, 2336–2347. DOI: 10.1021/acs.jpclett.9b03664.
8. Pavlo O. Dral, Bao-Xin Xue, Fuchun Ge, Yi-Fan Hou, MLatom: A Program Package for Quantum Chemical Research Assisted by Machine Learning. J. Comput. Chem. 2019, 40, 2339–2347. DOI: 10.1002/jcc.26004.
7. Pavlo O. Dral, MLatom: A Package for Atomistic Simulations with Machine Learning, (http://MLatom.com), 2013–2020.
6. Wen-Kai Chen, Xiang-Yang Liu, Weihai Fang, Pavlo O. Dral, Ganglong Cui, Deep Learning for Nonadiabatic Excited-State Dynamics. J. Phys. Chem. Lett. 2018, 9, 6702–6708. DOI: 10.1021/acs.jpclett.8b03026.
5. Pavlo O. Dral, Mario Barbatti, Walter Thiel, Nonadiabatic Excited-State Dynamics with Machine Learning. J. Phys. Chem. Lett. 2018, 9, 5660–5663. DOI: 10.1021/acs.jpclett.8b02469.
4. Pavlo O. Dral, Alec Owens, Sergei N. Yurchenko, Walter Thiel, Structure-Based Sampling and Self-Correcting Machine Learning for Accurate Calculations of Potential Energy Surfaces and Vibrational Levels. J. Chem. Phys. 2017, 146, 244108. DOI: 10.1063/1.4989536.
3. Raghunathan Ramakrishnan, Pavlo O. Dral, Matthias Rupp, O. Anatole von Lilienfeld, Big Data Meets Quantum Chemistry Approximations: The Δ-Machine Learning Approach. J. Chem. Theory Comput. 2015, 11, 2087–2096. DOI: 10.1021/acs.jctc.5b00099.
arXiv:1503.04987 [physics.chem-ph].
2. Pavlo O. Dral, O. Anatole von Lilienfeld, Walter Thiel, Machine Learning of Parameters for Accurate Semiempirical Quantum Chemical Calculations. J. Chem. Theory Comput. 2015, 11, 2120–2125. DOI: 10.1021/acs.jctc.5b00141.
1. Raghunathan Ramakrishnan, Pavlo O. Dral, Matthias Rupp, O. Anatole von Lilienfeld, Quantum Chemistry Structures and Properties of 134 Kilo Molecules. Sci. Data 2014, 1, 140022. DOI: 10.1038/sdata.2014.22.
Data set download link: figshare.